1. A 3 year old child with symptoms of stomatitis, gingivitis and dermatitis of open skin areas was delivered to a hospital. Examination revealed inherited disturbance of neutral amino acid transporting in the bowels. These symptoms were caused by the deficiency of the following vitamin:
2.
A 65 year old man suffering from gout complains of kidney pain. Ultrasound examination revealed renal calculi. The most probable cause of calculi formation is the strengthened concentration of the following substance:
3.
A patient consulted a doctor about symmetric dermatitis of open skin areas. It was found out that the patient lived mostly on cereals and ate too little meat, milk and eggs. What vitamin deficiency is the most evident?
4.
A 9-month-old infant is fed with artificial formulas with unbalanced vitamin B6 concentration. The infant presents with pellagral dermatitis, convulsions, anemia. Convulsion development might be caused by the disturbed formation of:
5. Cytoplasm of the myocytes contains a lot of dissolved metabolites resulting from glucose oxidation. Name the metabolite that turns directly into lactate:
6. On the ground of clinical presentations a patient was prescribed pyridoxal phosphate. This medication is recommended for correction of the following processes:
Explanation
Vitamin B6 (pyridoxine): converted to pyridoxal phosphate (PLP), a cofactor used in transamination (e.g. in ALT and AST), decarboxylation reactions, glycogen phosphorylase. Synthesis of cystathione, heme, niacin (Vit. B3), histamine and neurotransmitters including serotonin, epinephrine, norepinephrine, dopamine and GABA. Deficiency produces convulsions, hyperirritability, peripheral neuropathy (deficiency inducible by isoniazid and oral contraceptives), sideroblastic anemia due to impaired hemoglobin synthesis and iron excess.
7. You are studying functioning of a bacteria operon. The operator gene has been released from the repressor gene. Immediately after this the following process will start in the cell:
8.
Examination of a patient suffering from chronic hepatitis revealed a significant decrease in the synthesis and secretion of bile acids. What process will be mainly disturbed in the patient’s bowels?
9.
Products of some proteins hydrolysis and modification are the biologically active substances called hormones. Lipotropin, corticotropin, melanotropin and endorphins are synthesized in the hypophysis of the following protein:
10.
Nappies of a newborn have dark spots being the evidence of homogentisic acid formation. This is caused by the metabolic disorder of the following substance:
11. A 1,5-year-old child presents with both mental and physical lag, decolorizing of skin and hair, decrease in catecholamine concentration in blood. When a few drops of 5% solution of trichloroacetic iron had been added to the child’s urine it turned olive green. Such alteration are typical for the following pathology of the amino acid metabolism:
12.
A mother consulted a doctor about her 5-year-old child who develops erythemas, vesicular rash and skin itch under the influence of sun. Laboratory studies revealed decreased iron concentration in the blood serum, increased uroporphyrinogen I excretion with the urine. What is the most likely inherited pathology in this child?
13. When blood circulation in the damaged tissue is restored, then lactate accumulation comes to a stop and glucose consumption decelerates. These metabolic changes are caused by activation of the following process:
14.
Laboratory examination of a child revealed increased concentration of leucine, valine, isoleucine and their ketoderivatives in blood and urine. Urine smelt of maple syrup. This disease is characterized by the deficit of the following enzyme:
15.
A sportsman needs to improve his sporting results. He was recommended to take a preparation that contains carnitine. What process is activated the most by this compound?
16.
A patient complained about dizziness, memory impairment, periodical convulsions. It was revealed that these changes were caused by a product of decarboxylation of glutamic acid. Name this product:
17. A 35 year old man consulted a dentist about reduced density of dental tissue, high fragility of teeth during eating solid food. This patient suffers the most probably from the deficiency of the following mineral element:
18. A cerebral trauma caused increased ammonia generation. What amino acid participates in the excretion of ammonia from the cerebral tissue?
19.
A patient suffers from hepatocirrhosis. State of antitoxic liver function can be characterized by examination of the following substance excreted by urine:
20.
A clinic observes a 49 year old patient with significant prolongation of coagulation time, gastrointestinal hemorrhages, subcutaneous hematomas. These symptoms might be explained by the deficiency of the following vitamin:
21.
According to the model of double DNA helix that was suggested by Watson and Creek, it was established that one of chains would not be lost during replication and the second chain would be synthesized complementary to the first one. What way of replication is it?
22.
A patient underwent an operation on account of gall bladder excision that resulted in obstruction of Ca absorption through the bowels wall. What vitamin will stimulate this process?
23. A newborn child has convulsions that have been observed after prescription of vitamin B6 . This most probable cause of this effect is that vitamin B6 is a component of the following enzyme:
24.
A 46 year old woman suffering from chololithiasis developed jaundice. Her urine became dark-yellow and feces became colourless. Blood serum will have the highest concentration of the following substance:
Explanation
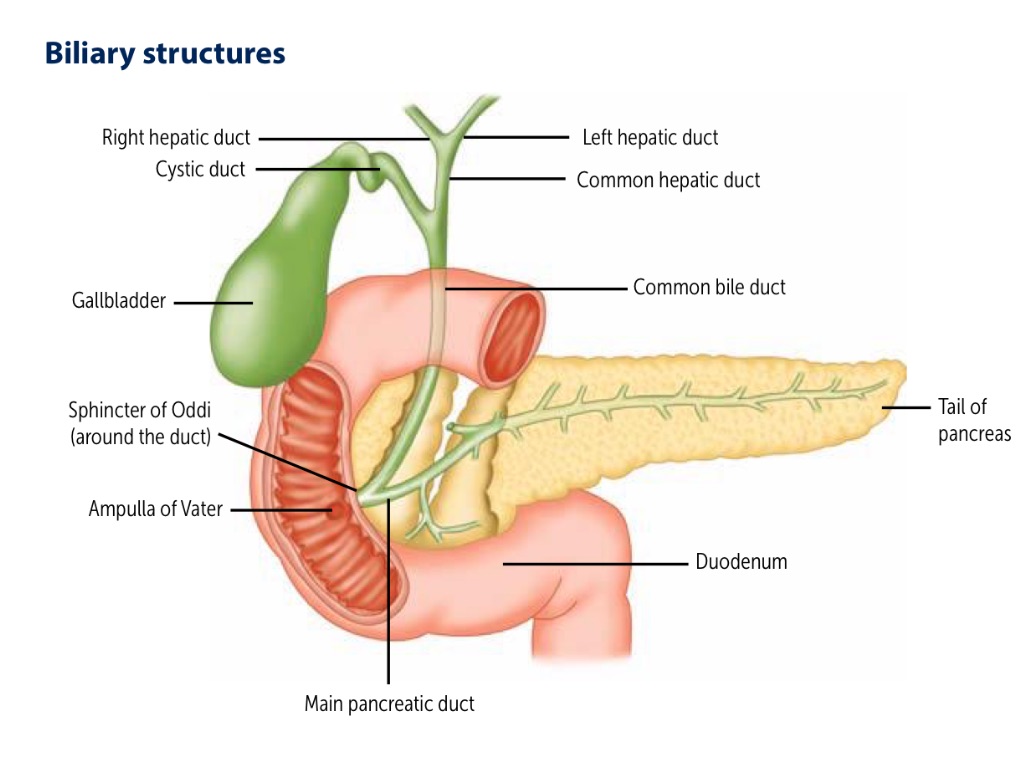
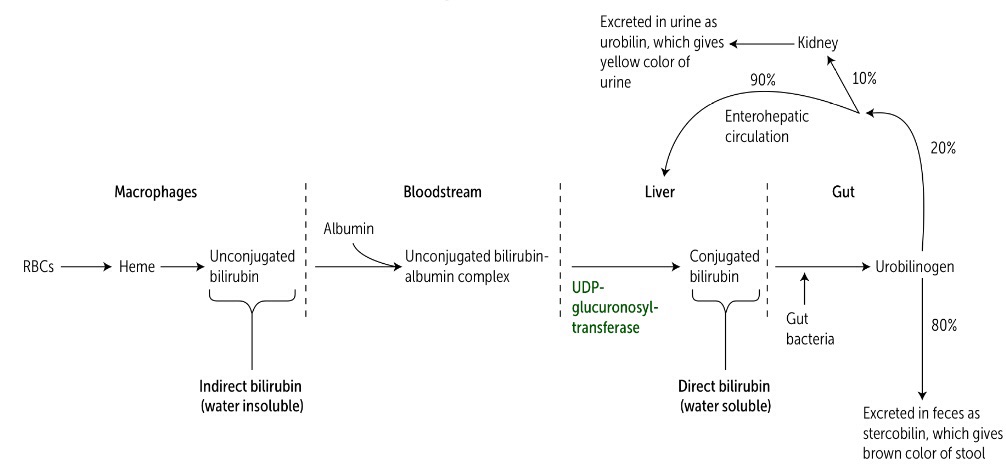
Cholelithiasis is obstruction of bile duct by a stone.
Obturation (obstruction, to close) of bile duct – it can be:
* Intrahepatic – blockage of intrahepatic bile ducts
* Extrahepatic – blockage of common bile duct (ductus choledochus).
Findings:
* malabsorption: bile salts do not enter the Small Intestine; no emulsification of fat.
*light coloured stool: due to lack of urobilin (which leads to lack of stercobilin).
*Jaundice (posthepatic, mechanic, obstructive): increased conjugated Bilirubin.
* Steatorrhea
The findings are specific for obstruction of bile duct and bile acid deficiency.
|
|
Indirect; Hemolytic; Prehepatic
|
Mixed; Parenchymal; Hepatic
|
Direct; Obstructive; Mechanic; Posthepatic
|
|
Stercobilin (faeces)
|
↑↑↑
|
Decreases (pale faces)
|
Absent (clay coloured faeces)
|
|
Type of bilirubin in blood
|
Unconjugated
|
Conjugated and Unconjugated
|
Conjugated
|
25.
A 48 year old patient complained about intense pain, slight swelling and reddening of skin over the joints, temperature rise up to 38oC . Blood analysis revealed high concentration of urates. This condition might be caused by disturbed metabolism of:
26.
Examination of a patient suffering from frequent hemorrhages in the inner organs and mucous membranes revealed proline and lysine being included in collagen fibers. Impairment of their hydroxylation is caused by lack of the following vitamin:
Explanation
Vitamin C (ascorbic acid): found in fruits and vegetables; an antioxidant; also facilitates iron absorption by reducing it to Fe2+ state. It is necessary for hydroxylation of proline and lysine in collagen synthesis; necessary for dopamine β-hydroxylase, which converts dopamine to norepinephrine. Deficiency leads to: scurvy – swollen gums, bruising, petechiae, hemarthrosis, anemia, poor wound healing, perifollicular and subperiosteal hemorrhages, “corkscrew” hair; Weakened immune response.
Type III collagen is found in blood vessels; Type IV collagen is found in basement membrane. Deficiency in Vitamin C disrupts the second stage of collagen synthesis in fibroblasts (hydroxylation of collagen) which results in petechiae, bruising, hemarthrosis.
27. Untrained people often have muscle pain after sprints as a result of lactate accumulation. This might be caused by intensification of the following biochemical process:
28. Examination of a patient revealed typical presentations of collagenosis. This pathology is characterized by increase of the following urine index:
29.
Labelled amino acids alanine and tryptophane were injected to a mouse in order to study localization of protein synthesis in its cells. The labelled amino acids will be accumulated near the following organellas:
30.
A full-term newborn child has yellowish skin and mucous membranes. This might be probably caused by temporary deficiency of the following enzyme:
31.
A patient has low rate of magnesium ions that are necessary for affixion of ribosomes to the endoplasmic reticulum. It is known that it causes disturbance of protein biosynthesis. At what stage is protein biosynthesis impaired?
32.
Blood of a 12 year old boy presents low concentration of uric acid and accumulation of xanthine and hypoxanthine. This child has genetic defect of the following enzyme:
33. Examination of a patient suffering from cancer of urinary bladder revealed high rate of serotonin and hydroxyanthranilic acid. It is caused by excess of the following amino acid in the organism:
Explanation
Serotonin, also called 5-hydroxytryptamine, is synthesized and stored at several sites in the body. It can be found in the intestinal mucosa, central nervous system and in platelets. Serotonin is synthesized from tryptophan, which is hydroxylated. The product, 5-hydroxytryptophan is decarboxylated to serotonin, which is also degraded by monoamine oxidase (MAO). Serotonin has multiple physiologic roles, including pain perception, affective disorders, and regulation of sleep, temperature and blood pressure.
3-hydroxyanthranilic acid is an intermediate in the metabolism of tryptophan.
34.
Study of conversion of a food colouring agent revealed that neutralization of this xenobiotic takes place only in one phase - microsomal oxidation. Name a component of this phase:
35.
A child has mental and physical retardation, grave damage of internal connective tissue. Urine analysis reveals keratan sulfates. What metabolic process is disturbed?
36.
A newborn child was found to have reduced intensity of sucking, frequent vomiting, hypotonia. Urine and blood exhibit increased concentration of citrulline. What metabolic process is disturbed?
37. On an electron micrograph a scientist has identified a structure formed by eight histone proteins and a part of DNA molecule which makes about 1,75 revolutions around the molecules. Which structure has been identified?
38.
A newborn develops dyspepsia after the milk feeding. When the milk is substituted by the glucose solution the dyspepsia symptoms disappear. The newborn has the subnormal activity of the following enzyme:
39. Some students developed myodynia after continuous physical activity during physical education. The reason for such condition was accumulation of lactic acid in the skeletal muscles. It was generated in the students’ bodies after activation of the following process:
40.
During starvation muscle proteins break up into free amino acids. These compounds will be the most probably involved into the following process:
41. A 1-year-old baby has been hospitalised for body and limbs lesions. Examination revealed carnitine deficiency in the child’s muscles. A biochemical reason for this pathology is the disorder of:
42. It was found out that some compounds, for instance fungi toxins and some antibiotics can inhibit activity of RNA-polymerase. What process will be disturbed in a cell in case of inhibition of this enzyme?
43. A patient complained about dizziness, memory impairment, periodical convulsions. It was revealed that these changes were caused by a product of decarboxylation of glutamic acid. Name this product:
44.
During hypersensitivity test a patient got subcutaneous injection of an antigen which caused reddening of skin, edema, pain as a result of histamine action. This biogenic amine is generated as a result of transformation of the following histidine amino acid:
45.
Vitamin B1 deficiency causes disturbance of oxidative decarboxylation of α-ketoglutaric acid. This leads to the impaired synthesis of the following coenzyme:
46.
Jaundice treatment involves administration of barbiturates inducing the synthesis of UDP-glucuronyl transferase. A medicinal effect is caused by the production of:
47.
It was revealed that T-lymphocytes were affected by HIV. Virus enzyme - reverse transcriptase (RNA-dependent DNA-polymerase) - catalyzes the synthesis of:
48. A patient has an increased pyruvate concentration in blood, most of it is excreted with the urine. What kind of avitaminosis has this patient?
49.
Urine analysis of a 12-year-old boy reveals high concentration of all aliphatic amino acids with the highest excretion of cystine and cysteine. US of kidneys revealed kidney concrements. What is the most likely pathology?
50. The greater amount of nitrogen is excreted from the organism in form of urea. Inhibition of urea synthesis and accumulation of ammonia in blood and tissues are induced by the decreased activity of the following liver enzyme:
51.
A doctor recommends a patient with duodenal ulcer to drink cabbage and potato juice after the therapy course. Which substances contained in these vegetables help to heal and prevent the ulcers?
52. A 46-year-old patient consulted a doctor complaining about joint pain that becomes stronger the day before the weather changes. Blood examination revealed an increased concentration of uric acid. The most probable cause of the disease is the intensified disintegration of the following substance:
53.
A patient has pellagra. Interrogation revealed that he had lived mostly on maize for a long time and eaten little meat. This disease had been caused by the deficit of the following substance in the maize:
54.
Researchers isolated 5 isoenzymic forms of lactate dehydrogenase from the human blood serum and studied their properties. What property indicates that the isoenzymic forms were isolated from the same enzyme?
55.
A 10-year-old girl has a history of repeated acute respiratory viral infection. After recovering she presents with multiple petechial hemorrhages on the sites of friction from clothing rubbing the skin. What kind of hypovitaminosis has this girl?
Explanation
Vitamin C (ascorbic acid): found in fruits and vegetables; an antioxidant; also facilitates iron absorption by reducing it to Fe2+ state. It is necessary for hydroxylation of proline and lysine in collagen synthesis; necessary for dopamine β-hydroxylase, which converts dopamine to norepinephrine. Deficiency leads to: scurvy – swollen gums, bruising, petechiae, hemarthrosis, anemia, poor wound healing, perifollicular and subperiosteal hemorrhages, “corkscrew” hair; Weakened immune response.
Type III collagen is found in blood vessels; Type IV collagen is found in basement membrane. Deficiency in Vitamin C disrupts the second stage of collagen synthesis in fibroblasts (hydroxylation of collagen) which results in petechiae, bruising, hemarthrosis.
56.
A patient presents with dysfunction of cerebral cortex accompanied by epileptic seizures. He has been administered a biogenic amine synthesized from glutamate and responsible for central inhibition. What substance is it?
57.
Osteolaterism is characterized by a decrease in collagen strength caused by much less intensive formation of cross-links in collagen fibrils. This phenomenon is caused by the low activity of the following enzyme:
58.
A patient has been diagnosed with alkaptonuria. Choose an enzyme whose deficiency can be the reason for this pathology:
59. Emotional stress causes activation of hormone-sensitive triglyceride lipase in the adipocytes. What secondary mediator takes part in this process?
60.
A 36 year old female patient has a history of collagen disease. Urine analysis is likely to reveal an increased concentration of the following metabolite:
61.
Vitamin A together with specific cytoreceptors penetrates through the nuclear membranes, induces transcription processes that stimulate growth and differentiation of cells. This biological function is realized by the following form of vitamin A:
62.
After severe viral hepatitis a 4 year old boy presents with vomiting, occasional loss of consciousness, convulsions. Blood test revealed hyperammoniemia. Such condition is caused by a disorder of the following biochemical hepatic process:
63.
Before the cells can utilize the glucose, it is first transported from the extracellular space through the plasmatic membrane inside them. This process is stimulated by the following hormone:
64.
A 2 year old child with mental and physical retardation has been delivered to a hospital. He presents with frequent vomiting after having meals. There is phenylpyruvic acid in urine. Which metabolism abnormality is the reason for this pathology?
65.
Examination of a child who hasn’t got fresh fruit and vegetables during winter revealed numerous subcutaneous hemorrhages, gingivitis, carious cavities in teeth. What vitamin combination should be prescribed in this case?
Explanation
Vitamin C (ascorbic acid): found in fruits and vegetables; an antioxidant; also facilitates iron absorption by reducing it to Fe2+ state. It is necessary for hydroxylation of proline and lysine in collagen synthesis; necessary for dopamine β-hydroxylase, which converts dopamine to norepinephrine. Deficiency leads to: scurvy – swollen gums, bruising, petechiae, hemarthrosis, anemia, poor wound healing, perifollicular and subperiosteal hemorrhages, “corkscrew” hair; Weakened immune response.
Type III collagen is found in blood vessels; Type IV collagen is found in basement membrane. Deficiency in Vitamin C disrupts the second stage of collagen synthesis in fibroblasts (hydroxylation of collagen) which results in petechiae, bruising, hemarthrosis.
Vitamin P (rutin) – permeability vitamin. They reduce permeability of blood vessels, especially capillaries. It prevents hyaluronic acid (the basic compound of the extracellular matrix) from degeneration by inhibition of the enzyme hyaluronidase. Ascorutin is a drug containing vitamin C and P; it is used to decrease the permeability of blood vessels.
66.
An experimental animal that was kept on protein-free diet developed fatty liver infiltration, in particular as a result of deficiency of methylating agents. This is caused by disturbed generation of the following metabolite:
67.
Characteristic sign of glycogenosis is muscle pain during physical work. Blood examination reveals usually hypoglycemia. This pathology is caused by congenital deficiency of the following enzyme:
68.
At the stage of translation in the rough endoplasmic reticulum, the ribosome moves along the mRNA. Amino acids are joined together by peptide bonds in a specific sequence, and thus polypeptide synthesis takes place. The sequence of amino acids in a polypeptide corresponds to the sequence of:
69. Children with Lesch-Nyhan syndrome have a severe form of hyperuricemia accompanied by the formation of tophi, urate calculi in the urinary tracts, as well as serious neuro-psychiatric disorders. The cause of this disease is the reduced activity of the following enzyme:
70.
The genetic defect of pyruvate carboxylase deficiency is the cause of delayed physical and mental development and early death in children. This defect is characterized by lacticemia, lactaciduria, disorder of a number of metabolic pathways. In particular, the following process is inhibited:
71.
An experiment proved that UV- irradiated skin cells of patients with xeroderma pigmentosum restore the native structure of DNA slower than the cells of healthy people due to the defect in repair enzyme. What enzyme takes part in this process?
72. One of the factors that cause obesity is the inhibition of fatty acids oxidation due to:
73.
For the study of serum proteins various physical and physicochemical methods can be used. In particular, serum albumins and globulins can be separated by this method:
74. Enzymatic jaundices are accompanied by abnormal activity of UDP-glucuronyl transferase. What compound is accumulated in blood serum in case of these pathologies?
75.
It is known that the monoamine oxidase (MAO) enzyme plays an important part in the metabolism of catecholamine neurotransmitters. In what way does the enzyme inactivate these neurotransmitters (norepinephrine, epinephrine, dopamine)?
76. A 20-year-old male patient complains of general weakness, rapid fatigability, irritability, decreased performance, bleeding gums, petechiae on the skin. What vitamin deficiency may be a cause of these changes?
Explanation
Vitamin C (ascorbic acid): found in fruits and vegetables; an antioxidant; also facilitates iron absorption by reducing it to Fe2+ state. It is necessary for hydroxylation of proline and lysine in collagen synthesis; necessary for dopamine β-hydroxylase, which converts dopamine to norepinephrine. Deficiency leads to: scurvy – swollen gums, bruising, petechiae, hemarthrosis, anemia, poor wound healing, perifollicular and subperiosteal hemorrhages, “corkscrew” hair; Weakened immune response.
Type III collagen is found in blood vessels; Type IV collagen is found in basement membrane. Deficiency in Vitamin C disrupts the second stage of collagen synthesis in fibroblasts (hydroxylation of collagen) which results in petechiae, bruising, hemarthrosis.
Vitamin B2 (riboflavin) deficiency – growth retardation, glossitis, conjunctivitis
Vitamin B1 (thiamine) deficiency – Beri-Beri (polyneuritis)
Vitamin A (retinol) deficiency – Night blindness
Vitamin B9 (folic acid) deficiency – macrocytic megaloblastic anemia
77.
By the decarboxylation of glutamate in the CNS an inhibitory mediator is formed. Name it:
78.
An unconscious patient was taken by ambulance to the hospital. On objective examination the patient was found to have no reflexes, periodical convulsions, irregular breathing. After laboratory examination the patient was diagnosed with hepatic coma. Disorders of the central nervous system develop due to the accumulation of the following metabolite:
Explanation
Substances absorbed into the bloodstream from the intestine pass through the liver, where toxins are normally removed. Many of these toxins (such as ammonia) are normal breakdown products of the digestion of protein. In hepatic encephalopathy (hepatic coma), toxins are not removed because liver function is impaired. Ammonia is produced by amino acid metabolism and intestinal urease-positive bacteria. In physiological conditions, it is mostly present as ammonium (NH4+) in serum. The urea or ornithine cycle, which is fully expressed in the liver exclusively, serves to converts NH4+ to urea prior to renal excretion and to maintain low serum concentrations. In hepatic coma, when the liver cannot remove toxins and urea cycle is not functional, all this occurs:
NH3 + α-ketoglutarate → Glutamate
α-ketoglutarate is used up which leads to:
· ↑glutamate → ↑GABA (inhibitory neurotransmitter)
· Inhibition of citric acid cycle/tricarboxylic acid cycle; this causes impairment of ATP formation.
· Inhibition of metabolism of amino acids (impairment of transamination reactions).
NH3 + Glutamate → Glutamine
Glutamine is an amide of glutamic acid which provides a non-toxic storage and transport form of ammonia (NH3). Ammonia increase synthesis of glutamine in brain. Accumulation of glutamine in brain results in elevation of osmotic pressure in nervous cells leading to brain edema.
NH3 + H+ → NH4+
In blood ammonia (NH3) is represented as ammonium ion (NH4+). Accumulation of ammonium ion impairs transport of ions (Na+, K+) through cell membranes and failure of transmission of nerve impulse.
Urea cycle takes place exclusively in the liver, so in hepatic coma, urea level is low. Glutamine toxicity in brain is dependent on increased ammonia concentration.
Bilirubin toxicity will most likely be related to increase hemolysis, which is not the case in this question. Histamine is a biogenic amine produced from the amino acid histidine.
79. A number of diseases can be diagnosed by evaluating activity of blood transaminases. What vitamin is one of cofactors of these enzymes?
80. Glycogen polysaccharide is synthesized from the active form of glucose. The immediate donor of glucose residues during the glycogenesis is:
81.
Hemoglobin catabolism results in release of iron which is transported to the bone marrow by a certain transfer protein and used again for the synthesis of hemoglobin. Specify this transfer protein:
82.
Pterin derivatives (aminopterin and methotrexate) are the inhibitors of dihydrofolate reductase, so that they inhibit the regeneration of tetrahydrofolic acid from dihydrofolate. These drugs inhibit the intermolecular transfer of monocarbon groups, thus suppressing the synthesis of the following polymer:
83.
A 28-year-old patient undergoing treatment in the pulmonological department has been diagnosed with pulmonary emphysema caused by splitting of alveolar septum by tissular tripsin. The disease is caused by the congenital deficiency of the following protein:
84.
A patient diagnosed with focal tuberculosis of the upper lobe of the right lung had been taking isoniazid as a part of combination therapy. After some time, the patient reported of muscle weakness, decreased skin sensitivity, blurred vision, impaired motor coordination. Which vitamin preparation should be used to address these phenomena?
85. Hepatitis B is diagnosed through laboratory tests that determine the presence of HBA-DNA in blood serum of the patient. What reference method is applied for this purpose?
86.
Nucleolar organizers of the 13-15, 21, 22 human chromosomes contain about 200 cluster genes that synthesize RNA. These regions of chromosomes bear the information on the following type of RNA:
87. Steatosis is caused by the accumulation of triacylglycerols in hepatocytes. One of the mechanisms of this disease development is a decrease in the utilization of VLDL neutral fat. What lipotropics prevent the development of steatosis?
Explanation
Hepatic steatosis can occur when humans are deprived of choline.
Choline + Phosphatidic acid → Phosphatidylcholine (lecithin, PC). In the liver PC can also be synthesized from phosphatidylserine (PS) and phosphatidylethanolamine (PE), when free choline levels are low, because it exports significant amounts of PC in bile and as a component of serum lipoproteins (needed for fat metabolism)
PS → PE →→→ PC. 3 methylation reactions between PE and PC. S-adenosylmethionine is the methyl group donor. If choline, phosphatidylcholine or methionine is deficient, there will be abnormal phospholipid synthesis, oxidative damage caused by mitochondrial dysfunction, lipoprotein secretion (remember, if VLDL cannot be secreted it will be accumulated & cause fatty liver degeneration as seen in hepatic steatosis). PC is also a major lipid component of lung surfactant.
Vitamin B12 is a cofactor for homocysteine methyltransferase and methylmalonyl-CoA mutase. Both vitamin B12 and B6 are needed in fatty acid (FA) metabolism. 88. Decarboxylation of glutamate
induces production of gamma-aminobutyric acid (GABA ) neurotransmitter. After breakdown, GABA is converted into a metabolite of the citric acid cycle, that is:
89.
Disruption of nerve fiber myelinogenesis causes neurological disorders and mental retardation. These symptoms are typical for hereditary and acquired alterations in the metabolism of:
90.
A child has a history of hepatomegaly, hypoglycemia, seizures, especially on an empty stomach and in stressful situations. The child is diagnosed with Gierke’s disease. This disease is caused by the genetic defect of the following enzyme:
91.
In case of alkaptonuria, homogentisic acid is excreted in urine in large amounts. The development of this disease is associated with a disorder of metabolism of the following amino acid:
92.
Patients with erythropoietic porphyria (Gunther’s disease) have teeth that fluoresce with bright red color when subjected to ultraviolet radiation; their skin is light-sensitive, urine is red-colored. What enzyme can cause this disease, when it is deficient?
93. A 49-year-old male patient with acute pancreatitis was likely to develop pancreatic necrosis, while active pancreatic proteases were absorbed into the blood stream and tissue proteins broke up. What protective factors of the body can inhibit these processes?
94.
According to the results of glucose tolerance test, the patient has no disorder of carbohydrate tolerance. Despite that, glucose is detected in the patients’ urine (5 mmol/l). The patient has been diagnosed with renal diabetes. What renal changes cause glucosuria in this case?
95.
A patient with hereditary hyperammonemia due to a disorder of ornithine cycle has developed secondary orotaciduria. The increased synthesis of orotic acid is caused by an increase in the following metabolite of ornithine cycle:
96.
A 39-year-old female patient with a history of diabetes was hospitalized in a precomatose state for diabetic ketoacidosis. This condition had been caused by an increase in the following metabolite level:
97. A patient with homogentisuria has signs of arthritis, ochronosis. In this case, the pain in the joints is associated with the deposition of:
98. Malaria is treated with structural analogs of vitamin B2 (riboflavin). These drugs disrupt the synthesis of the following enzymes in plasmodium:
99.
A 53-year-old male patient is diagnosed with Paget’s disease. The concentration of oxyproline in daily urine is sharply increased, which primarily means intensified disintegration of:
100. Cyanide is a poison that causes instant death of the organism. What enzymes found in mitochondria are affected by cyanide?
101. It has been found out that one of a pesticide components is sodium arsenate that blocks lipoic acid. Which enzyme activity is impaired by this pesticide?
102.
A 46-year-old female patient consulted a doctor about pain in the small joints of the upper and lower limbs. The joints are enlarged and shaped like thickened nodes. Serum test revealed an increase in urate concentration. This might be caused by a disorder in metabolism of:
103.
Those organisms which in the process of evolution failed to develop protection from H2O2 can exist only in anaerobic conditions. Which of the following enzymes can break hydrogen peroxide down?
104.
A patient has a critical impairment of protein, fat and hydrocarbon digestion. Most likely it has been caused by low secretion of the following digestive juice:
105. Prolonged fasting causes hypoglycemia which is amplified by alcohol consumption, as the following process is inhibited: